Researh
Identification of genes related with high regenerative ability
The capacity for regeneration varies greatly among metazoans, yet little is known about the evolutionary processes leading to such different regeneration abilities. In particular, highly regenerative species such as planarians and cnidarians can regenerate the whole body from an amputated fragment. By comparative genomics approach, we found genes conserved in the high regenerative species that were lost in the low-regeneration species during evolution. Furthermore, we observed regeneration failures in planarians after silencing the expression of the candidate genes. These observations shed insight into common genetic mechanism required for regeneration in metazoans, and may lead to practical applications for regenerative medicine in the future.

Duplication of dosage balanced genes by whole genome duplication
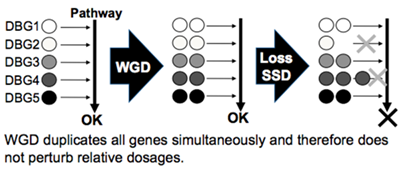
Although whole genome duplication (WGD) is often credited with great evolutionary importance, the processes governing the retention of these genes and their biological significance remain unclear. One increasingly popular hypothesis is that dosage balance constraints are a major determinant of duplicate gene retention. We test this hypothesis and show that WGD-duplicated genes (ohnologs) have rarely experienced subsequent small-scale duplication (SSD) and are also refractory to copy number variation (CNV) in human populations and are thus likely to be sensitive to relative quantities (i.e., they are dosage-balanced). By contrast, genes that have experienced SSD in the vertebrate lineage are more likely to also display CNV. This supports the hypothesis of biased retention of dosage-balanced genes after WGD. Dosage balance constraints simultaneously explain duplicate gene retention and essentiality after WGD.
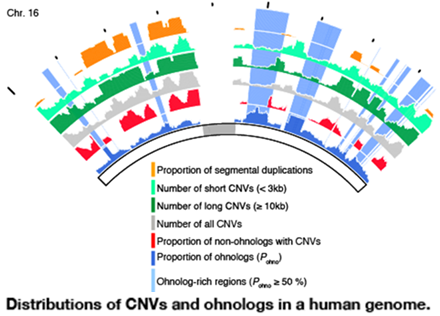
Copy number variations are distributed non-randomly in vertebrate genomes, however, it is unclear what genomic factors affect the deleterious effects of copy number variations and the biological significance of the biased genomic distribution of copy number variations remains poorly understood. We showed that non-ohnologs neighbouring ohnologs are unlikely to have copy number variations, resulting in ohnolog-rich regions in vertebrate genomes being copy number variation deserts. Our results suggest that the genomic location of ohnologs is a determining factor in the retention of copy number variations and that the dosage-balanced ohnologs are likely to cause the deleterious effects of copy number variations in these regions.
Application of whole genome duplication study to medical field
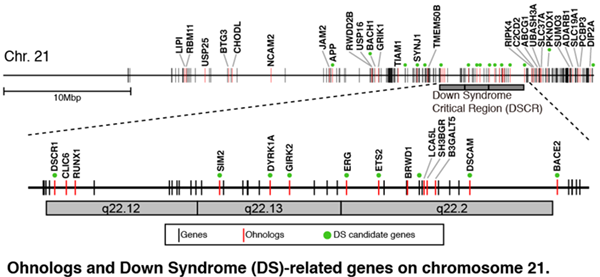
We also show that ohnologs have a strong association with human disease. In particular, Down Syndrome caused by trisomy 21 is widely assumed to be caused by dosage effects, and 75% of previously reported candidate genes for this syndrome are ohnologs which experienced no other copy number changes. We propose the remaining dosage-balanced ohnologs on chromosome 21 as candidate Down Syndrome genes. This clearly shows a persistent resistance to dose changes in genes duplicated by WGD. We also found that ohnologs are significantly overrepresented in genes mapping to pathogenic CNVs associated with developmental disorders and Alzheimer’s Disease. We propose that investigating copy number variation of ohnologs is an efficient means to find disease-related copy number variations.
Application of gene duplication study to ecological field
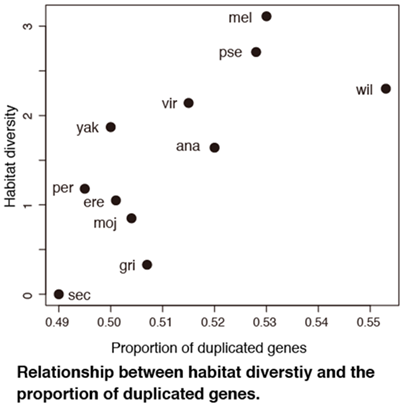
The mechanism by which genetic systems affect environmental adaptation is a focus of considerable attention in the fields of ecology, evolution, and conservation. However, the genomic characteristics that constrain adaptive evolution have remained unknown. A recent study showed that the proportion of duplicated genes in whole Drosophila genomes correlated with environmental variability within habitat, but it remains unclear whether the correlation is observed even in vertebrates whose genomes including a large number of duplicated genes generated by whole-genome duplication (WGD). Here, we focus on fully sequenced mammalian genomes that experienced WGD in early vertebrate lineages and show that the proportion of small-scale duplication (SSD) genes in the genome, but not that of WGD genes, is significantly correlated with habitat variability. Moreover, species with low habitat variability have a higher proportion of lost duplicated genes, particularly SSD genes, than those with high habitat variability. These results indicate that species that inhabit variable environments may maintain more SSD genes in their genomes and suggest that SSD genes are important for adapting to novel environments and surviving environmental changes. These insights may be applied to predicting invasive and endangered species.
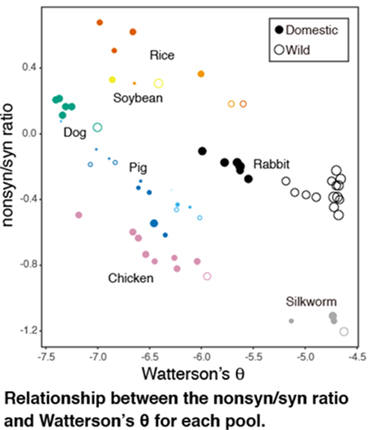
Deleterious genetic variation in domestic animals and plants
A fraction of genetic variants segregating in any population are deleterious, which negatively impacts individual fitness. The domestication of animals and plants is associated with population bottlenecks and artificial selection, which are predicted to increase the proportion of deleterious variants. However, the extent to which this is a general feature of domestic species is unclear. Here, we examine the effects of domestication on the prevalence of deleterious variation using pooled whole-genome resequencing data from five domestic animal species (dog, pig, rabbit, chicken, and silkworm) and two domestic plant species (rice and soybean) compared with their wild ancestors. We find significantly reduced genetic variation and increased proportion of nonsynonymous amino acid changes in all but one of the domestic species. These differences are observable across a range of allele frequencies, both common and rare. We find proportionally more single nucleotide polymorphisms in highly conserved elements in domestic species and a tendency for domestic species to harbor a higher proportion of changes classified as damaging. Our findings most likely reflect an increased incidence of deleterious variants in domestic species, which is most likely attributable to population bottlenecks that lead to a reduction in the efficacy of selection. An exception to this pattern is displayed by European domestic pigs, which do not show traces of a strong population bottleneck and probably continued to exchange genes with wild boar populations after domestication. The results presented here indicate that an elevated proportion of deleterious variants is a common, but not ubiquitous, feature of domestic species.
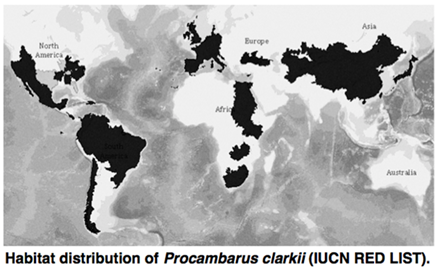
Genomic analyses of Procambarus clarkii
Wild Procambarus clarkii is distributed in the southern United States and north-eastern Mexico. P. clarkii has been globally introduced for domestic consumption and as food for fishes and frogs, and the species is established in northern America, southern America, Europe, Africa, and Asia. The introduced P. clarkii colonize rapidly, even though the introduction effect on the crayfish is extremely low. P. clarkii is one of the best organisms for investigations of genomic architectures linked to invasiveness and rapid adaptation.